Datos: PubChem CID 33741 — National Library of Medicine
Efectos
Analgesia moderadaeuforia levenáuseamareosudoraciónriesgo de convulsiones (dosis-dependiente)estreñimientodependencia con uso prolongado.
Mecanismo de acción
Mecanismo dual: agonista débil del receptor mu-opioide (μ) e inhibidor de la recaptación de serotonina y noradrenalina (IRSN). Su metabolito activo O-desmetiltramadol (M1, formado por CYP2D6) tiene afinidad 200 veces mayor por el receptor mu que el compuesto padre. El componente monoaminérgico contribuye a la analgesia y puede causar síndrome serotoninérgico en combinación con otros serotoninérgicos.
Vida media
Vida media de eliminación de 5-7 horas para el tramadol y 7-9 horas para su metabolito activo O-desmetiltramadol (M1). La vida media puede prolongarse en insuficiencia renal o hepática.
Toxicidad
IDENTIFICACIÓN Y USO: El tramadol es un polvo cristalino blanco, inodoro, con sabor amargo. El tramadol es un analgésico opioide que está indicado para el manejo del dolor moderado a moderadamente severo en adultos. En medicina veterinaria, puede utilizarse para el tratamiento del dolor postoperatorio o crónico o la tos. EXPOSICIÓN HUMANA Y TOXICIDAD: Las manifestaciones de sobredosis son similares a las de otros agonistas opiáceos e incluyen depresión respiratoria, letargo, flacidez del músculo esquelético, coma, convulsión, bradicardia, hipotensión, paro cardíaco, miosis, vómito, piel fría y húmeda, colapso cardíaco y muerte. Se han reportado reacciones anafilactoides graves y raramente fatales. Otras reacciones de hipersensibilidad reportadas incluyen prurito, urticaria, angioedema, broncoespasmo, necrólisis epidérmica tóxica y síndrome de Stevens-Johnson. El síndrome serotoninérgico potencialmente mortal puede ocurrir con tramadol, particularmente con el uso concomitante de otros fármacos serotoninérgicos, fármacos que deterioran el metabolismo de la serotonina (p. ej., inhibidores de la MAO), o fármacos que deterioran el metabolismo del tramadol (p. ej., inhibidores de las isoenzimas 2D6 y 3A4 del citocromo P-450 [CYP]). El uso de tramadol se ha asociado con un riesgo aumentado de hiponatremia que requiere hospitalización. Un estudio sugiere un riesgo moderadamente aumentado de un efecto teratogénico del tramadol. Cuando se usa tramadol durante el embarazo, existe un riesgo grave de síndrome de abstinencia neonatal. ESTUDIOS EN ANIMALES: Hubo 910 exposiciones a tramadol como agente único reportadas al ASPCA Animal Poison Control Center durante 2009-2013. De los 749 perros, 142 fueron sintomáticos. Los síntomas incluyeron sedación/letargo, vómito, taquicardia, vocalización o ataxia, y agitación o temblores. De los 157 gatos, 96 fueron sintomáticos con síntomas tales como midriasis, hipersalivación, letargo, ataxia o taquicardia, y vómito. Las ratas con serotonina reducida estuvieron predispuestas a convulsiones inducidas por tramadol, y las concentraciones de serotonina se asociaron negativamente
Farmacología
Opioide atípico con mecanismo dual: agonismo mu débil e inhibición de recaptación de serotonina y noradrenalina. Analgésico de potencia intermedia indicado para dolor moderado. Riesgo de convulsiones (dosis-dependiente) y síndrome serotoninérgico. Su metabolito M1 (vía CYP2D6) es el principal responsable del efecto opioide. Metabolizadores ultrarrápidos tienen mayor riesgo de toxicidad.
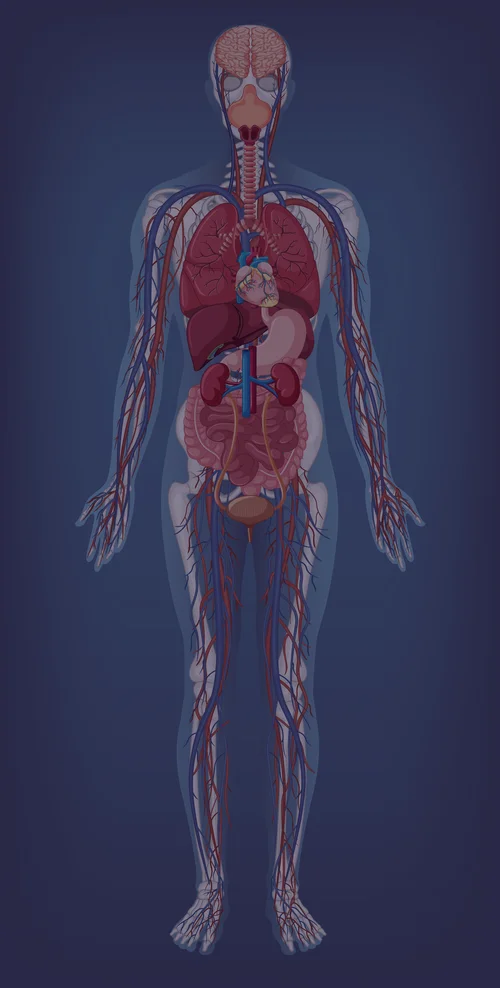
Efectos en el organismo
Cerebro
Pulmones
Corazón
Hígado
Riñones
Tracto GI
Órgano afectado
Efecto primario
No afectado
Farmacocinética
Vida media
Vida media de eliminación de 5-7 horas para el tramadol y 7-9 horas para su metabolito activo O-desmetiltramadol (M1)
Tiempo al pico
1-2 horas (oral)
Duración
4-6 horas
Inicio
30-60 minutos (oral)
Biodisponibilidad
68-72% (oral)
Metabolismo
Hígado (CYP2D6 → O-desmetiltramadol, CYP3A4)
Excreción
Renal (90%)
Vol. distribución
Volumen de distribución de 2.5-3.0 L/kg. Biodisponibilidad oral del 68-72%. Unión a proteínas plasmáticas del 20%. Metabolismo hepático vía CYP2D6 al metabolito activo O-desmetiltramadol.
Cronología farmacocinética
Inicio30-60 minutos (oral)
Pico1-2 horas (oral)
Duración4-6 horas
Evidencia regulatoria · FDA
Evidencia FDA
FAERS · openFDA labels
Datos regulatorios y de farmacovigilancia de la FDA (EE.UU.): reportes espontáneos de eventos adversos (FAERS) y secciones estructuradas del prospecto oficial. FAERS refleja frecuencia de REPORTE, no causalidad.
Frecuencia de REPORTE, no causalidad. FAERS es un sistema pasivo: sesgo de sub-notificación y de mediatización.
openFDA · Etiqueta oficial
Prospecto estructurado (FDA)
openFDA · Structured Product Labelling
No hay prospecto FDA registrado para esta sustancia (no aprobada por FDA como medicamento).
Riesgos para la salud — Calculadora de dosis
Riesgo base ARES46 · Moderado
Componentes ARES
T · Toxicidad
51
D · Dependencia
44
M · Mortalidad
45
A · Abuso
46
Herramienta educativa no personalizada: no estima una dosis segura y no incorpora pureza, tolerancia, interacciones, salud individual ni adulteración. Los rangos se conservan como datos metodológicos auditados hasta completar curación por fuente primaria.
0.00 mgSin efecto
Arrastra la bolita
25 mg
50 mg
100 mg
200 mg
400 mg
Cardiovascular
Corto plazo
Hipotensión ortostáticaPosible prolongación QT a dosis altas
Los rangos de dosificación fueron compilados a partir de Goodman & Gilman's (14ª ed.), Rang & Dale (9ª ed.) y publicaciones indexadas en PubMed. Los valores representan promedios poblacionales para la vía de administración principal y no consideran variabilidad individual, tolerancia, peso corporal ni interacciones.
Perfil bioquímico · ChEMBL
Afinidad a receptores in vitro
EMBL-EBI ChEMBL · CC-BY-SA 3.0
Datos experimentales de bioactividad publicados en literatura y curados por ChEMBL. Las afinidades (Ki, Kd, IC50, EC50) son mediciones in vitro; no equivalen a efectos clínicos.